
Translating Non-Uniform RBE Modeling to the Clinic
A Concept... this time explained in more depth.

Version2: Based on comments and feedback, I’d added some additional context to the document. I think in shortening, I may have lost too much. This attempt will be a more complete discussion of my rationale.
Premise:
We cannot/should not move protons from 1.1 RBE compared to photons to some “better” modeling of this effect without clinical validation. Protons have been using a basic conversion (1.1) since they went clinically live. We are on the cusp of “thinking” we have better calculations. These new approaches demand clinical validation.
A recent article was published evaluating Mayo Clinic’s variable RBE model. As many people know, when we deal with particles there is a bit more to consider when trying to model dose. To me, this is a very detailed and complex area - quite foreign to many of us who have lived in the photon world, so today, we’ll try to keep it rather simple.
Just a few quotes from the paper for flavor.
Main results: Marked differences were observed between the results of the phenomenological proton RBE models, as reported in previous studies…
Significance: The study highlights the importance of considering cell-specific characteristics and detailed radiation quality information for accurate RBE calculations in proton therapy…
So this article is finding that how we model RBE changes within the beam seems to depend on cell-specific parameters and that within 6 different current models - the results have significant variance.
We know that we have been too simplistic in our approach. (Ref - TG256). Below is an image showing the measured absorbed dose in a beam treating a target from 10cm to 20cm depth. Below you can see in the red curves how the "layers” are given to create the cumulative SOBP (spread out Bragg peak) dose curve.

But the EFFECT of that dose, we believe varies. Below is RBE within a passive scattered beam profile WITHIN the 95% SOBP dose. This shows that over a 10 cm span - from 10 cm to 20 cm of depth, the RBE changes based on our best information.
This is from TG-256 showing the effective dose will vary from the proximal to the distal end of the 95% range by approximately 30%.

So within the therapeutic dose of a passive scatter beam (the flat “top” dose), the distal end of the SOBP is potentially running at an RBE of 1.3 or higher. And if you read farther down in the TG 256 report, you will see that RBE can be as high at 1.7 in the distal fall off region (so beyond the 95% isodose line - ie in the region where absorbed dose really falls off. If you’ll notice, to create a SOBP, most of the dose is delivered to the far distal end of the target, then layered on this dose are much smaller amounts of dose to “smooth” the top - it is likely where this primary distal end is stopping where the largest differences in effect are seen).
This is, in part, why I wrote last year about the requirement for randomized trials. Click Here
Below are images from a uniform scanning prostate plan so that you can be clear on what we are discussing - I actually think it is far easier to discuss in the setting of passive approaches rather than in the pencil beam setting - but the same principles hold up - some we think to a lesser degree and some might be amplified.
First, a “composite” 2 beam initial plan. The red contour of the prostate, with a red 98% line. The dark blue is 95% and the cyan is the 60% isodose line. Note the “over shooting” laterally due to range uncertainty - we literally add 5mm beyond the PTV laterally due to range uncertainty (so ant / post the ultimate PTV margin is 4mm, but laterally the ultimate PTV margin is 9mm for example). Here, it doesn’t seem to matter - that excess dose still lands in nothing important (it seems) and it ensures coverage due to things that we can’t predict.

Below is a single beam view showing the same thing but just for one beam.

Again, what you “see” above is based on an RBE of 1.1 across the entirety of the beam dose. In contrast, what our reference physics document states, is that between the blue lines of the lower, single beam view - is that you should multiply that dose by the values in Figure 1 as you move from left to right.
So for the above example, there might be an area at the distal end of the 95% that really is closer to 1.3 *0.95 = 1.235. 23% hot. Remember, this is only one beam so the “actual” overdose might be in 11%-12% range.
Ok - remember that and we’ll resume discussing pieces of the larger puzzle.
As mentioned above, we have traditionally converted dose with an RBE of 1.1 but computer planning systems keep getting more advanced and we are beginning to better factor in additional factors of LET and/or variable RBE modeling. The Mayo clinic paper discusses their approach. I won’t dive into the paper, we’ll stay at a higher / broader level today.
I think it is safe to say, that we are certain that our traditional modeling is too simplistic - that by integrating LET and better RBE modeling that we can further optimize dose distributions and better define tumor control and risk to any adjacent organs.
There are, in fact, a number of different models and a number of approaches to this issue (this study evaluated 6 different approaches to give you a feel for the complexity), but as a physician on the pure clinical side of the equation, I always return to the basic question of:
How do we make the jump to the clinical implementation of these models?
Maybe it is simple and we just use lab and physics studies without any clinical data and simply jump?
That would be similar to the jump from homogeneous calcs to heterogeneous calcs. We “thought we knew” enough to make the math / physics jump and we did it, but here, I think the physical science pairs with cellular RBE type effects. Together, those interactions make this transition have at least one extra layer of uncertainty (likely far more).
Today, we’ll try to propose a safe, practical jump within the clinic that I believe could be used to validate any transition between models with rather limited patients and rather quickly (say 100 patients, within 2 years).
Background Context:
In simple terms, the components to this conversation can be highly technical. We’ll try to really simplify.
Three components come into play:
1) Uncertainty.
Particles have uncertainty in their behavior - we model them with Monte Carlo calculations for this reason (thousands of calcs to estimate the likely landing spot for the individual particles). And where they ultimately stop and deposit energy affects RBE for that tissue on a cellular level.
2) LET.
Linear Energy Transfer describes the transfer of energy based on things we know pretty well - for example, the energy of the protons, the density of the tissue, the atomic composition of the tissue, and beam characteristics like width and shape. Since we know most of these things, we’re better at modeling this one than…
3) RBE.
Relative Biological Effectiveness is the relative effective difference between particles and photons - ie, when they deposit dose (which they do differently in part due to the charge and mass), there is a relative difference in cellular effect. It varies based on a number of factors - again exactly how the treatment damages the DNA - rate of double strand breaks vs. single strand breaks and density of that damage.
This attribute is affected by the particle, your biological endpoint (cell survival vs tissue damage), tissue sensitivity due to cell type, fractionation scheme and dose rate. So this one corresponds to LET but is harder to calculate.
All of that said, today’s models simply use 1.1 to convert between proton and photon doses. Protons have 1.1x the impact so we “give” ~10% less dose for the same effect.
That is as simple as I can get it. They are all intertwined and there are thousands of people working on “resolving” as much of this science as we can in the lab and translating that information into planning systems. But ultimately, what I believe we should care about is:
Does an improvement in the model - moving from non-LET optimized, uniform 1.1 RBE to more complex models result in better outcomes? - ie a verification of the work to outcomes.
As a reference document, I recommend this AAPM TG-256 document. As a physics document goes, it is quite readable and is my personal go-to refresher.
Is there any clinical data supporting under representation of distal dose?
Yes - this is all pretty well established.
While, in general, I think protons are very safe and have been used to push dose higher for some really high risk cancers adjacent to critical structures, I think there are examples where these issues might have been appreciated as clinically important. First, there is an older experience in peds cases where simple beam arrangements attempting to stop the beam just in front of critical structures without feathering of the distal range resulted in necrosis. More recently, I think the MSKCC head and neck series showing maybe a higher risk of osteoradionecrosis of the mandible right where the beams stop is once again consistent with an under modeling of dose effects at distal beam edge (posterior oblique arrangements essentially stopping in the mandible). Finally, if there is an increased risk of rib fracture in breast cases, this would be supporting rationale.
Again, these are all mitigatable issues once you consider the known underpinnings of our planning approaches, but I think each demonstrate a real clinical outcome risk if we put too much emphasis on just what the plan shows us.
In my current clinic, we really, really try to avoid stopping “just before” a critical structure. When we have to, we add multiple beams and even for the beam in question, we’ll overshoot the critical target for part of the delivered dose and then back the beam range up even shorter - essentially “moving the junction” to counter / minimize any of these unknowns. This is simply a known limitation.
(I honestly think it is going to be smaller effect size than some of the TG256 values from a clinical impact - I think whenever doses get to be represented in really small volumes, they have far less effect but that is more opinion.)
My stance therefore is that we should mandate clinical verification for at least some sights.
My analogy: A Spaceship
Consider all the crazy math that goes into rocket ships. It is amazing what we can do, but seeing the rocket take off and now land is what truly validates the work. While there is complex math and physic principles underlying the lunar landing, seeing it happen drove it all home. Here to, I believe we must validate the work.
Five Years Ago:
Five years ago I jumped from a photon world into the world of protons. New eyes on a new approach. And I was honestly shocked at how big of a volume that we treated for prostate cancer. If you look at Figure 2 above and focus on the blue 95% line compared to the red prostate contour, it is a crazy big volume coming from current IMRT planning - all of that “excess” lateral dose.
So I started reading and studying trying to make sense of the approach and you quickly get to range uncertainty. I get that and that makes sense - pair it with an area in which you have “too many physicists” and you get very conservative answers - answers that I would just say would have never allowed the progress that Dr. Timmerman discussed in his jump to SBRT approaches (full article: The World According to Timmerman)
But while the “range uncertainty” makes sense, it is undermined by this increasing RBE across the target. Again, in the reference document stating a number of 1.7 for regions beyond the 95% distal end. (So take the 60% line and multiply that by 1.7 and get something like full dose at the “60% isodose” line - again, you would cut it in half moving the effective two beam dose from 60% to 80% but still, the plan might be 20% off for small regions spanning a few mms (in figure 3 above, the distance between the 95% and 60% line is a little less than 5mm as an example - and the 1.7 referenced in TG256 might be a fraction of a mm of that - maybe).
So back then I was surprised that I didn’t see more clinical work to answer this uncertainty. My conceived approach was quite simple, we should basically REMOVE the range uncertainty from our beams for prostate cancer which would then SHIFT the higher dose RBE uncertainty into the target. If the curves above are correct with higher effective doses at the distal end of the SOBP, we would potentially see no difference. Or perhaps, even improvement in outcomes.
I struggled to get buy-in. It was a leap too far and realistically, moving to a uniform scanning center in 2019 was a decade too late for this type of work. (maybe)
But the idea remained, somewhere deep down.
How non-uniform models are being implemented today:
First off, I have no first hand clinical experience, but in general, here is my understanding of how these new models are being implemented into the clinic.
They are being used to “increase” the accuracy of the dose modeling. i.e. they are showing more of this distal dose and so by showing the extra dose and optimizing that dose into the target, you are able to reduce the dose delivered to the patient. Which then reduces dose to the adjacent OARs. So “total dose to the patient” is lowered, “tumor effective dose” remains “constant” thereby lowering OAR doses.
I think of them as stepwise improvements. Current uniform representations are where we are today. The next step is LET-optimized approaches. (As stated above, these are things we know pretty well and applying this relates to a better approximation of RBE than current but still not modeling all known variables). And finally, non-uniform RBE modeling with LET-optimized dose deposition modeling. At least that is my simple construct.
But validating these models within our current application framework is nearly an impossible task. Your hope is that you pick up a decrease in a very uncommon toxicity - at best. The integral dose reduction (compared to IMRT) is laughable and we can’t even clearly demonstrate those differences. The path we are on creates a result towards never verifying the outcome with clinical validation of the model.
What about PET or MRI as a measurement tool to validate the model?
Yep. We are getting better and better with determining where the dose is deposited. The direct interaction of the beam with the nuclei of targets can be measured with PET (REF). So no radiotracers - the protons hit nuclei and then they create B+ decay emitters with half-lives of 20 and 2 min. Granted, this helps with range, but it doesn’t include any interactions with electrons and it does NOT measure the main pathway for protons to deposit energy: via electromagnetic interactions.
Kind of crazy but we can measure where the protons collide into the nuclei of atoms with PET imaging post treatment. The closer the time to the delivery of the beam, the more precise the information. We simply work in the fanciest field in all of medicine. An amazing field!!
MRI is another approach being explored, but it seems a bit farther off. (REF)
And while PET is a rather direct measurement of part of the dose - go back up to the definitions of LET vs. RBE and see if you think this is really more along the lines of LET measurement or RBE measurement. It doesn’t tell us anything about how cell lines react or differences in survival curves for cancer control vs. complication risk. It is closer to what we want to validate, but it still isn’t there - at least that is my read.
So we still need at least one strong clinical example that what we do in the lab with cell lines and radiation dose, translates to clinical outcomes.
If I’m missing puzzle pieces or have a gap in my understanding, please reach out or comment.
My Simple Idea: A path to clinical validation.
Like I said, we tend to use these “improved” models to demonstrate that we can “put more of the delivered dose” into the target and therefore, decrease the total delivered dose to the patient and thereby potentially reduce toxicities.
But realistically, this approach leads to - at best - non-inferiority approaches HOPING to see a toxicity fall from 4% to 2-3% for example. And with time, toxicities fall. We simply get better. There is almost no way that we will ever document that these models - unique from the general progress of out field - actually do what we think they are doing in a clinical setting that I see as feasible.
Instead, I think there is real opportunity to approach the problem from the opposite direction. Implement the model and keep the dose delivered to the patient the same and track tumor control outcomes via kinetics and demonstrate that, yes, what we are delivering is completely consistent with a higher effective clinical dose.
And for that, we turn to prostate cancer. A cancer where we literally have decades of PSA kinetic data and where we KNOW where the steep shoulder of the dose response curve lies. In an incredibly ironic twist, we can use prostate cancer treated with protons to help push the treatments with proton therapy towards a new clinically validated RBE non-uniform modeling world.
The Concept:
Use PSA kinetics, which are amazingly good predictors of long-term cure rates, to assist in finding clinical validation of the models.
Treat intermediate risk prostate cancer. Treat men to 6000 cGy(RBE)/20 fractions with either our current “traditional model” or a new “LET/RBE optimized model”. Note: within the new model, the dose to the patient needs to remain the same, but use the model, not to reduce the dose to normal OARs, but to increase the deposition of dose within the target. So in this scenario, we might push the mean target dose from 6000 to 6400 using the optimized non-linear model.
Then simply monitor PSA kinetics and toxicity across a cohort of the two approaches. My thinking is that we know 60/20 is the “low end” of the appropriate dose curve - shorter than traditional, but still fractionated to where I trust the kinetics data.
If we “optimize” the dose delivery and actually bump it up 5%-10% within the prostate due to improvements in modeling - it has a real chance to show up in improved PSA kinetics at the 1 yr and 2 yr mark - using the simple benchmark of “lower to faster”. (I’ve discussed kinetics and the data supporting this simple concept extensively on this site - suffice it to say, there is a plethora of data).
What we should not see with the change is worse kinetics or increases in toxicity. If we see either, we need to stop and pause. If we keep the delivered dose to the patient similar, the risk is very low - after all, it is same amount of dose leaving the machine, just an improvement in laying it down in the most appropriate / accurate fashion.
My Data: A “Use Case Example”
I’ve tracked data in my clinic since I’ve arrived and I have over 300 patients where I’ve followed PSA kinetics post proton therapy. Single MD, highly uniform from a contouring and planning aspect and I feel pretty confident that within the first 100 patients (50 treated with current modeling and 50 treated with adjusted modeling) that you would likely see a difference in kinetics IF you truly altered the dose. I think I see that type of slight change in my data.
Since my arrival back in 2019, I’ve treated with two primary approaches. I started at 7920 RBE(cGy) / 44 fractions because every prostate before my arrival was treated that way (aside from an SBRT proton trial cohort) and I literally had no proton experience - so I did what they did.
About 8-10 months in, I began moving towards a fractionation based on great results out of the University of Florida Proton Therapy Institute program (REF). 7250 RBE (cGy) / 29 fractions. I have a much smaller cohort of SBRT that I’ve discussed at length but realistically, I have two main cohorts.
They aren’t perfectly matched - today the 7920 group probably has larger prostates and a bit worse AUA but they are very similar in that, for one year I did all one way, and now I do 95%+ via the shorter approach. And the shorter approach is a bit hotter based on EQD2 calcs - here are two side by side with the 7920 on top and the 7250 shown below:

And kinetics - here are the resulting PSA kinetics for the two cohorts - the shorter follow-up arm is the 7250 cohort.
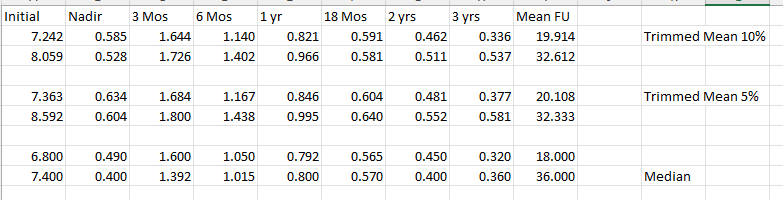
I could dress it up more to “make a point”, but the data is the data. Initial PSA slightly favor the 7250 arm and median follow-up is about 1 yr different - this table includes over 170 patients. Obviously still a young dataset, but... Medians carry the least “information” regarding the cohort and are basically the same. Trimmed mean carry much data from the cohort. Very few fail (~120 men in the 7250 arm with 1 current failure - but it is a PSA>10 at 1 yr), so reasonable to exclude them - trimmed means do that.
And in summary, kinetics seem to slightly favor 7250 over 7920. Not dramatically or to any massive degree, but they appear faster to lower. That is my use case. By 1-2 years, with likely around 100 patients (50 per arm), I think you will be AT LEAST able to CLAIM a clinical validation of your non-uniform modeling. This trend is completely consistent with the history of radiation in the treatment of prostate cancer. Stronger results from higher doses with better long-term outcomes and PSAs that reach lower levels faster.
Why this approach?
First, I believe that we require validation.
Secondly, it is an amazingly low risk approach - nearly a guarantee of no harm.
Third, it has real potential to demonstrate not just a safe transition of basic proton dose modeling calculation but an ability to document via kinetics that we have likely IMPROVED our dose representation.
Forth, I can’t think of another setting where this type of difference is even feasible within the next 2 years.
We have a large volume daily patient base of prostate cancer for accrual.
Tear it up / break it down, or let’s get it done.
What if my thinking on kinetics is wrong and even if we adjust the dose “properly” we don’t see differences in outcomes.
I think this is a real possibility. Frankly our results in prostate cancer have gotten so very strong that we might not pick up the dose change, but if not here, then where?
In general, data strongly supports that faster to lower is better, but there isn’t a ton of data using this approach out there. I get that, but what we should not see in the 100 patients is a significantly different impact on either toxicity or higher / worse kinetics within a moderate fractionated approach. I think if you see either of those, then we need to be cautious that we are shifting more than we believe we are.
And I understand that this might throw off a false negative reading, but I’d rather it be here in this setting than buried within clinical improvements and stage migration and general technology improvements within a high risk / reward cohort. A move from our traditional modeling approach, at least to me, is too critical to jump to without some type of confirmatory end-to-end clinical validation that what we see in the lab, appears to translate directly.
And I’d rather err on the side of caution.
I believe we need something easy to complete, relatively quick and accruable, and repeatable:
These models are coming. There are multiple approaches out there. Maybe they are all just simply better but somehow, I think they should be validated or AT LEAST CLOSELY monitored. And watching for toxicity events that happen at a 2% clip within a non-inferiority trial structure is far too insensitive. Here we use basically the entire population to study effects - a magnitude or 10 stronger to evaluate our changes.
We likely can’t validate all of the components - like RBE based on tissue and cell line type and cure vs. complication rate, but I do think it is wise for us to demonstrate some method to validate the jump from the lab to the clinic.
The consideration of moving to a new dose model for the treatment of a child, while pushing dose constraints and OARs limits, makes me convinced that clinical validation in a lower risk cohort is a required pre-requisite.
And using this approach, every model should be able to acquire 100 prostate cases - easily. Maybe it isn’t perfect and maybe the kinetics can’t really show “improvement” in the model but AT LEAST there will be some validation of the jump to the new model. I think the proton industry nearly requires that of us.
Granted, I believe we largely jumped in photons from homogeneous to non-homogeneous plans without this type of validation but here are some stats showing the massive scale that favors photons in picking up warning / safety issues:
As of 2021, less than 280,000 patients had been treated with protons in the history of the technology. Compared to 2020 when 1.06 million patients were treated with photon radiation in the US alone.
The scale on the photon side adds safety and the jump to heterogeneous was rather straightforward and “obvious” - photons transmit through bone and lung differently. Here, the issue is far more complex and difficult to comprehend. I purposefully left the exercise within the realm of passive scanning approaches where it is pretty straightforward - the mental complexity within PBS is a magnitude more obtuse - at least to me.
And even in the prior scenario where we used to treat lung like bone, I ran duplicate plans slowly transitioning my clinical practice for years - literally. I think we should treat this transition of modeling with an additional level of caution and respect due to complex interactions within radiobiological effect and linear energy transfer. Today, in many sites, we push dose closer to the edge than we have ever done in the history of our field. I believe we owe this type of validation to this approach - to both our past and future patients.
The lack of randomized data hurt the field now 30 years into our history. We need to avoid running various different models at different institutions. That approach will harm the technology moving forward in the long-run. I think it is wise for us to consolidate around mechanisms where we can validate clinical outcomes for these laboratory based improvements as we shift to non-uniform RBE modeling.
If you look at carbon data, even the basic prescriptions out of Japan and the EU have historically been different. It would be a shame for our proton modeling to de-coalesce into a similar non-uniform structure.
And yes, I see the irony that prostate cancer might be used to validate progress in proton therapy dosimetry, but maybe, that is why it is so appropriate. After all, they aren’t going away and we have plenty of them under beam - if they can help push the dose calculations forward, that would be a path towards better.
A sincere thank you to those who reached out or commented based on the first release. It has been something I have considered for half a decade and I think my first attempt over-simplified my thinking. I hope this one is more clear. Whether it is correct as the best path, I’m less convinced, but I’m certain it is an area where we need to have more discussions about how we progress from A»B.
As always, thank you for the support and for reading along.
Amazing thoughtful insightful article. ASTRO (and ACRO) should have you directly involved in Proton committee leadership.
Sorry, but I'm still trying to get clarity on exactly what the prescriptions would be.
>>>
within the new model, the dose to the patient needs to remain the same, but use the model, not to reduce the dose to normal OARs, but to increase the deposition of dose within the target. So in this scenario, we might push the mean target dose from 6000 to 6400 using the optimized non-linear model.
>>>
If the physical/absorbed (not BED) doses don't change, the only thing that could be different would be LET optimisation (how the physical dose got to the voxel).
If you are saying "the dose to normal/OAR should stay the same", and if we can assume that they won't be near the Bragg Peak, then it's both the physical dose and the BED staying the same for normal/OAR. You would either have to have a "better plan" where you keep the Normal/OAR but get more physical dose to the tumour, or you've done LET (and RBE) optimisation (which is a better plan under the assumption being tested).
If you are allowing the mean target dose to go as high as 6400cGy(RBE), then it's an LET/RBE optimised plan with a loose upper constraint on mean dose (BED) to tumour (>6000 cGy(RBE), weak or no constraint up to 6400 cGy(RBE)) with a fixed upper bound on on the OAR/Normal to match non LET/RBE optimised plans. Maybe I'm getting tripped up on the original statement of the prescription (
("6000 cGy(RBE)" rather than "minimum of 6000 cGy(RBE) but iso-toxic with the non-optimised plans".
Although they may not be taking the data, aren't people kind of already doing this with LET optimised plans (they also aren't calculating the BED based on those LET optimisations)? They might be doing this with the idea that they are avoiding placing the high LET dose in OAR, but when looking at sample plans, I always see the high LET region being put *in* the tumour.
I think the (probably too) bold move would be to do an LET/RBE optimised plan but keep the mean dose (BED) to tumour to see if there is lower toxicity. I guess it depends: What is the bigger clinical issue right now, lack of success in control or too high toxicity? If (big if) there's great control and low toxicity, it might be close to Pareto optimal already.